
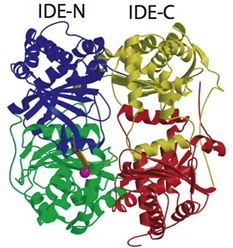
1. Structure and function of human insulin degrading enzyme (IDE) as well as therapeutic potential of IDE.
Insulin degrading enzyme (IDE) is a zinc metalloprotease and it is involved in the clearance of insulin and amyloid β, peptides involved in the progression of diabetes and Alzheimer's disease, respectively. IDE has unusual substrate selectivity that represents unique properties in protein-protein interaction. We solved structures of human IDE in complex with four substrates (insulin B chain, amyloid-beta (1-40) (Aβ), amylin, and glucagon) to elucidate the molecular basis of this unique substrate selectivity. Our structure model also provides a valuable tool in starting the exploration of IDE-based therapeutics in controlling cerebral Aβ accumulation and blood sugar levels. Future directions in studying IDE in my lab include the elucidation of the molecular basis in the regulation of IDE catalysis, the discovery of small molecule IDE activator and inhibitor leads, and the protein engineering of IDE to make an IDE-based therapeutic for Alzheimer's disease.
2. Structure and functions of bacterial toxins secreted by human bacterial pathogens.
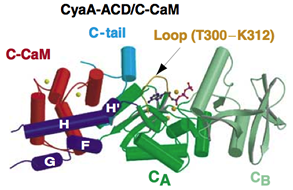
One toxin family that we have studied is adenylyl cyclase toxins which by themselves are not active till they enter into target cells and are associated with cellular proteins that serve as the activator. These adenylyl cyclase toxins become highly active to raise the intracellular cyclic AMP (cAMP) of its host cells to pathogenic levels. The unregulated increase of intracellular cAMP level can alter the functions of host cells to benefit the bacterial propagation. We have studied two adenylyl cyclase toxins, edema factor (EF) secreted by bacteria that cause anthrax and CyaA, which is secreted bacteria that cause whooping cough. Both EF and CyaA bind the cellular calcium sensor, calmodulin, with high affinity. We have solved the X-ray crystal structures of EF and CyaA as well as applied biochemical and biophysical analyses to address how calmodulin binds and activates EF and CyaA.
EF has an N-terminal 30 kDa PA binding domain followed by a 43 kDa catalytic core and a 17 kD helical domain. The extended form of calmodulin initiates the contact with the helical domain of EF which subsequently results in the insertion of calmodulin between the interface between the catalytic core and helical domain of EF. The binding of calmodulin stabilizes a catalytic loop that is crucial for catalysis. Such mechanism differs from the removal of autoinhibition observed in many CaM targets such as MLCK and the induced dimerization found in calcium-dependent K+ channel. EF does not share sequence and structure homology with mammalian adenylyl cyclases. However, EF also utilizes two-metal-ion catalytic mechanism which is also shared by many enzymes that perform the phosphoryl transfer reaction, such as mammalian adenylyl cyclase, DNA polymerase, and RNA polymerase. Thus, our studies has not only substantially improved our understanding in how calmodulin binds and activates its effectors, but also provides an example for the evolutionary drive of structural diverse enzymes to have the conserved mechanism of catalysis. To further address how calmodulin binds to EF and how EF converts ATP to the reaction products, cAMP and PPi, we want to continue using structural and biochemical approaches to construct a molecular movie that can best describe this interesting biological process.
We will continue these approaches to elucidate the principles in how protein-protein interaction leads to catalytic activation as well as how two proteins from two different organisms evolve to gain the desired biological activities. Many bacterial toxins, such as botulinum toxin (BoTox) and Cholera toxin, have been developed as the experimental and therapeutic tools. We also are exploring the therapeutic potential of adenylyl cyclase toxins in human diseases as well. We are also interested in better understanding in the regulation of exotoxins that contain adenylyl cyclase activity. Such toxin has also been identified from Pseudomonas aeruginosa which is one of hospital-acquired pathogens that threatens the health of the immuno-compromised patients such as those with AIDS or cystic fibrosis (ExoY). Genomic sequences of Yersinia pestis (plague) and Yersinia pseudotuberculosis (gastrointestinal syndromes) reveal a new member of adenylyl cyclase toxin that is associated with Tcc gene. This suggests that adenylyl cyclase toxin may be used broadly by pathogenic bacteria to alter the host defense. Although both CyaA and ExoY share sequence identify with EF (28-35%), they differ significantly from EF in their regulation. ExoY can not be activated by calmodulin but it can be activated by the unknown host cellular factor. CyaA is activated by calmodulin but the structure requirement of CyaA for the activation by calmodulin differs drastically from that of EF. We want to identify the activator of ExoY and address how CyaA and ExoY are activated by the biochemical and structural analyses.
3. The characterization and optimization of inhibitor leads of anthrax toxins.
The incident of bioterrorism-related anthrax in 2001 has moved the challenge against anthrax from an obscure agricultural problem to the center of bio-defense. Giving the ease of making antibiotic-resistant anthrax strains and unknown enemies, the best defense against anthrax is to build up a battery of possible antidotes against anthrax. We have already developed several small molecular anti-anthrax toxin leads that can potently inhibit the action of anthrax toxins, EF and lethal factor. Drugs of low molecular mass have been the major workhorses in the pharmaceutical industry to cure human diseases. Small molecular weight drugs are easier to deliver than peptides, and could enter into host cells to specifically inhibit the action of EF and LF inside the cells. We have discovered several leads that can specifically compete the binding of EF to substrate, among those, the most potent EF inhibitor is an approved drug, adefovir that can selectively inhibit the activity of EF both in test tube (27 nM Ki) and in cultured cells (IC50 around 100 nM). Adefovir is an acyclic nucleoside and it is approved to treat chronic hepatitis B virus infection. In addition, we have used the combination of cell-based screen and surface plasmon resonance analysis to screen a 10,000 compound library which results in the identification of a lead compound (NO2-10506-2A) that can block the interaction between EF and CaM with 10 µM affinity. Toward finding the LF inhibitor, we have worked with Milan Mrksich to develop a mass spectrometry-based high throughput screen to identify a lead that can specifically inhibit LF with 1 μM Ki (DS-998).
Our anti-anthrax toxin research now moves into the new phase that is to optimize the leads. Since the pharmaceutical industries have not moved into this arena due to the unpredictability in the usage of anthrax as the bioterrorism agent, I have chosen to work with the academic scientists to further improve the potency of small molecule anti-toxin. To improve the affinity of adefovir to EF, we collaborate with Jan Florián at Loyola University, Roger Johnson at SUNY, and Brian Shoichet at UCSF as well as Gilead Sciences Inc. to perform quantitative structure-activity relationship (QSAR) analysis and structure determination of EF-inhibitor complex. From this data, we will use quantum mechanic/molecular mechanic (QM/MM) analysis to construct the molecular model. Based on this model, we will synthesize second and third generation analogs to progressively improve the affinity of analogs to EF. Both the affinities of NO2-10506-2A to EF and DS-998 to LF are in low μM range. We will work with Milan Mrksich to use iterations of chemical synthesis, structure determination, as well as molecular modeling to understand how these leads bind to anthrax toxins and to improve the affinity of NO2-10506-2A to EF and DS-998 to LF.
We will also develop suitable animal models for drug target validation. I have worked with Steve Leppla at NIH to develop mouse model challenged with edema toxin (EF+PA). We have found that edema toxin causes a plethora of pathologic effects shortly after administration followed by death within three days. We are in the process of testing whether adefovir alleviate the illness caused by edema toxin. We will work with Michéle Mock at Pasteur Institute to study the effect of adefovir on animal models challenged with anthrax spores. Such experiment is vital as the proof-of-principle experiment to validate EF as a viable target against anthrax infection. We will also work with both Michéle Mock and Steve Leppla to test the analogs of NO2-10506-2A and DS-998 that have low nM affinity to EF and LF, respectively. The development and testing of anti-anthrax toxins inhibitor will not only provide crucial experimental tools to address the role of these toxins in anthrax pathogenesis, but also serve as the lead for therapeutics in fighting against infectious diseases such as anthrax, whooping cough, 10-20% of nosocomial infections, and potentially plague.
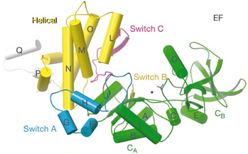
4. Structural basis for calmodulin-mediated regulation of mammalian small conductance potassium channel.